What methods do you recommend for RNA isolation?
We recommend a column-based method, including:
- Norgen Biotek Total RNA Purification Kit
- Zymo Research Quick-RNA™ Kits
- Arcturus PicoPure® RNA Isolation Kit
- Ambion PureLink® RNA Mini Kit
- Qiagen RNeasy Kits
For FFPE RNA isolation, we recommend a kit designed for FFPE samples, including:
- Norgen Biotek FFPE RNA Purification Kit
- Zymo Research Quick-RNA™ FFPE Kit
- Arcturus® Paradise® PLUS FFPE RNA Isolation Kit
- PureLink™ FFPE RNA Isolation Kit
- Qiagen RNeasy FFPE Kit
Organic methods such as TRIzol® Reagent should be subsequently followed with a column-based clean-up method. Isolation methods that capture RNAs shorter than 200 nt may result in increased 5S and 5.8S rRNA. Do not use carrier RNA during isolation.
Can I use TRIzol® or other phenol-chloroform based extractions for RNA isolation?
We do not recommend the use of TRIzol or similar methods as any carryover of organic solvents may inhibit downstream enzyme activity. If using TRIzol extracted RNA, we recommend using a column-based purification of the RNA prior to input into the kit.
Can I use carrier RNA during RNA isolation?
We do not recommend the use of carriers during RNA isolation. If a carrier is required, please contact Tecan NGS Technical Support for more information.
Can I use Ovation SoLo RNA-Seq System with RNA from any organism?
The Ovation SoLo RNA-Seq System should be suitable for total RNA input from any organism. For information on organism specific rRNA or targeted transcript depletion contact Tecan NGS Technical Support.
Do I need to use high-quality total RNA?
When using with the Ovation SoLo RNA-Seq System, samples should be of high molecular weight with little or no evidence of degradation. While it is impossible to guarantee the highest levels of performance when using RNA of lower quality, this system should allow the successful analysis of somewhat degraded samples. With such samples, users may experience lower yields and may encounter affected sequencing metrics.
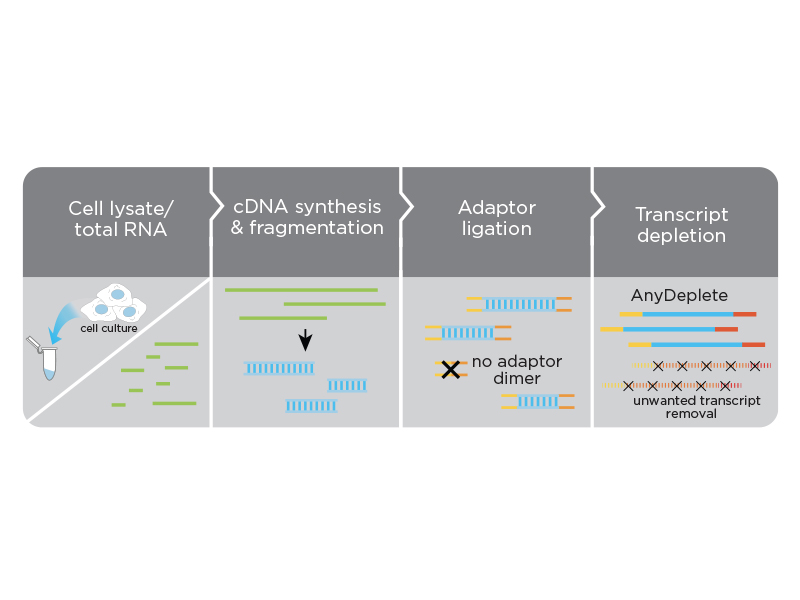
Do I need to perform an rRNA depletion or poly(A) enrichment step before processing samples with the Ovation SoLo RNA-Seq System?
No. The system is designed to use total RNA as input. rRNA depletion or poly(A) enrichment are not necessary. rRNA, as well as other transcript types, can be targeted for depletion using the AnyDeplete technology embedded in the workflow.
Can I sort my cells into PBS or another buffer prior to beginning the cell lysis protocol?
For best results we recommend sorting cells directly into the Lysis Buffer Master Mix prepared with the kit, or resuspending cell pellets in the Lysis Buffer Master Mix. Other buffers (e.g. sheath fluid) may inhibit reactions in the kit and yield variable sequencing results. It is recommended for other buffers not to exceed 1 ul. Contact Tecan NGS Technical Support for additional information.
What cell types are compatible with the lysis buffer provided with the kit?
The lysis buffer has been tested with mammalian cell inputs up to 500 cells and should be compatible with most dissociated cell types. Some direct cell inputs may need to be optimized depending on cell type. Decreased performance may be observed with cell types containing excessive lipids, minerals, collagen, etc. or a cell wall, or with very large cells. Non-dissociated cells, tissue, or LCM samples may require additional optimization of the workflow.
Can contaminating genomic DNA interfere with Ovation SoLo RNA-Seq System performance?
Yes, contaminating genomic DNA may be incorporated into libraries. For this reason DNase treatment is incorporated into both the Cell Lysis and Total RNA Input workflows.